Introduction
This report presents the work I carried out to introduce a new component into the muDock codebase.
The complete implementation is available in muDock PR #105.
Context
As explained in section 2 of the muDock vectorization paper, the general context of this work is drug discovery: the process through which candidate molecules are investigated as potential effective treatments for a disease. One of the initial phases of this process is virtual screening, where computational techniques are used to analyse a large set of candidate molecules and select the most promising ones before experimental validation in a laboratory. Virtual screening provides an initial filtering step, reducing the amount of work that must later be performed in the laboratory and therefore reducing time and costs.
Molecular docking is one of the techniques used in virtual screening. Given a receptor, typically a protein, and a ligand, meaning a candidate molecule, the objective is to estimate how the ligand may arrange itself inside an interaction region of the protein called the binding site. The quality, and therefore the probability, of an arrangement is evaluated through an energy term used as a proxy for binding affinity. A single spatial arrangement of the ligand is called a pose and includes the position, orientation, and internal torsions of the molecule.
To compare poses, docking uses a scoring function: a function that assigns a numerical value to each configuration, approximating the quality of the interaction between the ligand and the receptor. The task of docking is therefore to explore the pose space and identify the poses associated with the most favourable scores.
The computational problem is divided into two levels. The first concerns the number of candidate molecules to evaluate, which can generally be very large. The second concerns the number of possible poses to explore for each receptor-ligand pair. The degrees of freedom of the complex come from the ligand being able to move in space and rotate internally. In general, an exhaustive search over the pose space is not practical.
For this reason, docking engines usually use heuristics and optimisation algorithms that seek a compromise between computational cost and solution quality. In addition, many evaluations of different poses or molecules can be performed independently, making molecular docking a perfect use case for HPC.
Objective
The objective of the project was to implement a scoring function in C++ by using the existing infrastructure and pipeline of muDock. In particular, the implemented scoring function is Vinardo, described in the original paper. This required adapting the data representation, atom typing, preprocessing of atom pairs, and score computation to the existing components.
The main difficulty was therefore not only implementing the scoring function formula, but integrating it into an already structured codebase. The work required understanding the muDock architecture, comparing the behaviour with smina as the reference implementation, and introducing all the infrastructure required around the scoring kernel: atom typing, PDBQT topology information, pair preprocessing, and data management within the pipeline.
Project output
At the end of the project, the produced output was:
- C++ code integrated into the codebase;
- an execution pipeline for both scoring-only and scoring with a genetic algorithm;
- functional tests against the reference implementation.
Related Work and Background
This section explains at a high level the muDock architecture and the essential components for docking. It then explains the theoretical operation of Vinardo.
muDock architecture
muDock was created as a microapp for molecular docking, built to replicate an AutoDock-like workflow. In addition to providing a docking engine, the codebase is designed as an environment for experimenting with optimisation and porting techniques, such as vectorisation and support for different execution backends.
In the context of this work, the most relevant aspect is the deliberate separation between the phases of the pipeline: input parsing, molecule representation, scoring, search, and output production. This organisation makes it possible to modify a single component, such as the scoring function, without rewriting the entire execution flow.
At a high level, the execution flow is the following:
- the paths of the protein and ligand files are passed to the executable;
- the protein is parsed and loaded into memory as stable data because it is used to evaluate every ligand under analysis;
- ligands are progressively read, parsed, and inserted into a work queue;
- workers consume ligands from the queue, group them into batches, and execute the configured pipeline, which may consist of one or more stages;
- results are written into the ligand properties and produced as output.
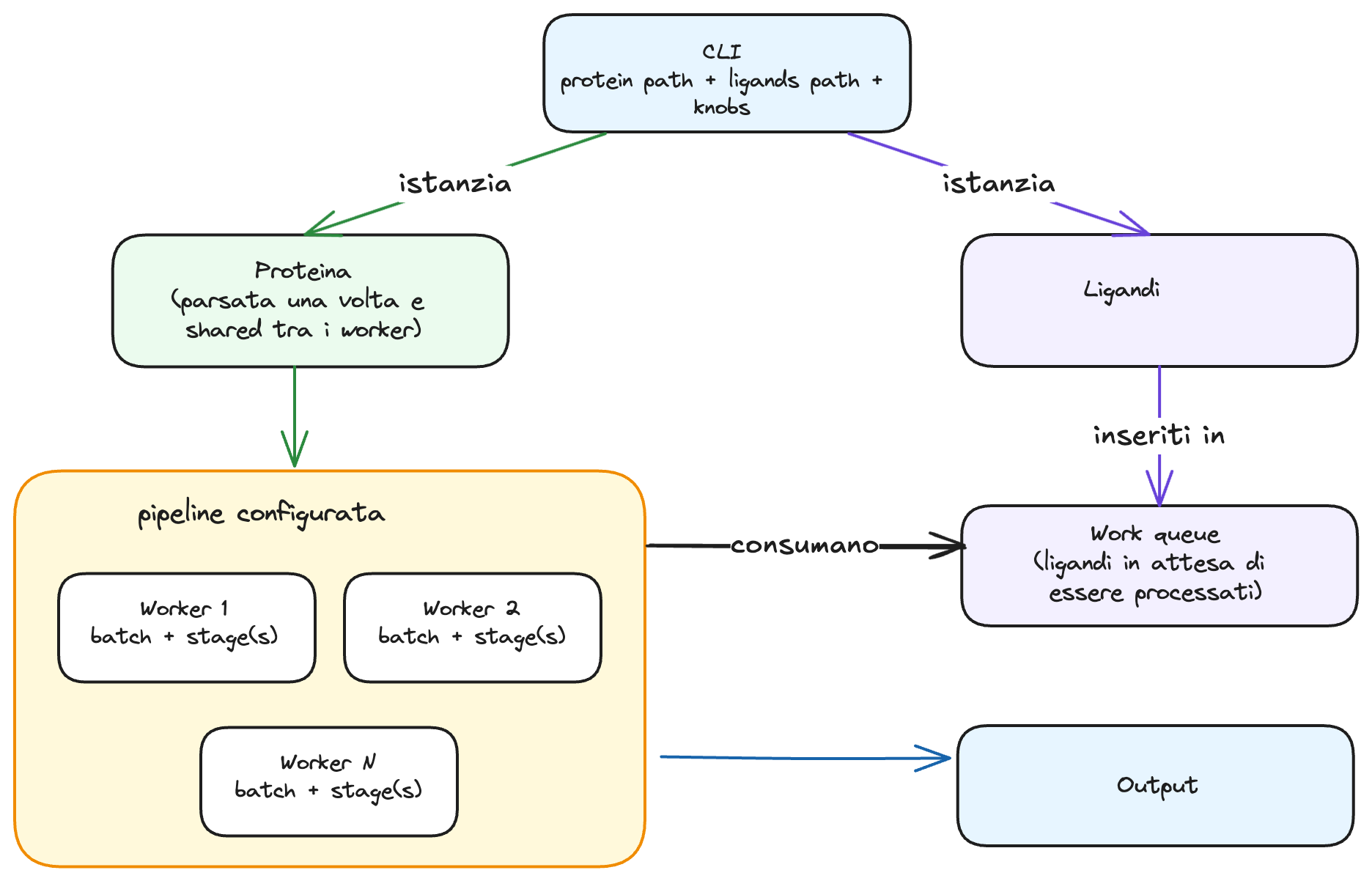
In general, a pipeline consists of one or more stages, where each stage represents a unit of work in the pipeline. In muDock, the stages relevant to this work are those related to scoring and genetic optimisation. Each stage follows a common cycle:
prepare: prepares the data required to execute the stage on the current batch;operator(): performs the actual work;teardown: collects the results and writes them into the output data structures.
This structure can be seen as a stateful functor. The object maintains the state required for execution, such as references to buffers and scratchpads, and makes the computation invocable through operator().
For example, the scoring stage can be represented as follows:
template<typename queue_type>
struct scoring_stage {
std::shared_ptr<scratchpad<queue_type>> scratch;
void prepare(batch<static_molecule>& batch);
void operator()();
void teardown(batch<static_molecule>& batch);
};The available pipelines include a scoring-only pipeline, which calculates the score associated with the interaction between the protein and ligand in the given configuration, and a genetic pipeline, where the genetic stage uses the scoring function to evaluate the poses generated during the heuristic exploration of the pose space. In the second case, the parameters called knobs in the codebase control the number of individuals, number of generations, mutation probability, and other aspects of the search.
At this point it is necessary to introduce the abstractions used to manage memory and data because this context requires enough flexibility to work independently from the execution backend and the framework used on that backend.
muDock mainly uses two abstractions: buffer and scratchpad. A buffer represents a typed memory area that can be used to read and write data, but also to move it between the host, usually the machine launching the computation, and the device, the machine used to perform the intensive computation, for example one equipped with a GPU.
The scratchpad is a shared registry of buffers. It allows stages to retrieve the data they need through common keys, avoiding unnecessary reconstruction or copying. The scratchpad can contain data that remains stable during the run, such as the representation of the protein, as well as data produced by one stage and consumed by the next. One example is the buffers associated with poses, containing ligand coordinates, which are produced during genetic exploration and then used by the scoring stage to evaluate the generated configurations.
The fact that stages can pass data through shared buffers and key conventions is fundamental to guarantee stable contracts between different stages and compose them flexibly. This becomes clearer later, when the integration of the Vinardo-based scoring stage only requires implementing a stage that respects the input and output contracts, without modifying the genetic pipeline or the other existing scoring stages.
The fundamental component of each stage is the kernel, which represents the actual numerical computation. The kernel is invoked through operator() and works on data already prepared by the previous phase. Since this is an HPC context and different execution backends must be supported, there can be different implementations of the same kernel, for example a CUDA version for GPU execution and a standard C++ version for CPU execution. For this work, the generic interface of the Vinardo kernel was implemented but specialised only for the CPU, leaving a GPU version as a possible future development.
Vinardo scoring function
Vinardo was developed as an evolution of the AutoDock Vina scoring function. For this reason, it preserves many of its characteristics and the same general structure. The quality of a pose is evaluated through a scoring function that sums energy contributions calculated over pairs of atoms.
Before describing the scoring function, it is useful to distinguish between the conformation-dependent score, meaning the spatial arrangement, and the affinity value reported as an estimate of binding free energy. In the Vina model inherited by Vinardo, the conformation-dependent score is denoted by and depends on the specific ligand pose.
In particular, consists of an intermolecular contribution associated with protein-ligand interactions and an intramolecular contribution associated with interactions internal to the ligand:
The value of is calculated as the sum of the contributions over the atom pairs considered valid:
where represents the set of atom pairs selected as valid during preprocessing. The considered pairs are:
- protein-ligand pairs, obtained from the Cartesian product between the ligand atoms and protein atoms;
- ligand-ligand pairs, selected only when they satisfy the required topological conditions.
In both cases, pairs containing at least one hydrogen atom are discarded. For ligand-ligand pairs, only atom pairs separated by more than three covalent bonds and able to move relative to each other are retained.
The contribution associated with a single atom pair must then be defined. In the previous formula, and represent the atom types of the two atoms, while is the surface distance between the atoms in the pair. It is obtained by subtracting the radii associated with the two atom types from the Euclidean distance between the atomic centres:
The function consists of multiple weighted energy terms:
The Gauss and Repulsion terms are evaluated for all pairs, while Hydrophobic and HBond depend on the atom types involved. In particular, the hydrophobic term is active only for pairs of hydrophobic atoms, while the hydrogen bond term is active when the pair may represent a donor-acceptor interaction.
The individual terms are defined as follows:
From an implementation perspective, the Hydrophobic and HBond terms are enabled through two flags precomputed during preprocessing. This means that scoring does not need to reevaluate the atomic properties of the pair and only has to apply the function to the prepared data.
The weights used by Vinardo are those reported in the original paper:
The main parameters of the terms are:
Once the conformation score has been calculated for the poses of a ligand, it is converted into affinity through a function , producing an estimate of the binding free energy.
For the best pose:
For subsequent poses, Vina instead subtracts the intramolecular contribution of the best pose to preserve the relative ranking:
The Vina paper also proposes a concrete form for , which was later inherited by Vinardo. The function normalises the value with respect to the number of rotatable bonds in the ligand:
Operationally, refers to the number of rotatable bonds in the ligand. This value is not retrieved directly from the field in the PDBQT file, but reconstructed from an in-memory topological representation of the ligand structure. In particular, the rotors described by the PDBQT torsion tree are considered. A rotatable bond is counted only when both endpoint atoms are not hydrogens and each has at least two bonds with non-hydrogen atoms.
From an implementation perspective, this structure therefore requires several components: atom typing compatible with Vinardo, a data representation suitable for scoring, a preprocessing phase to construct valid atom pairs, and a scoring function.
Implementation
This section describes the components implemented to integrate the new scoring function into the existing pipeline. The description follows the data flow: from the topological information extracted while parsing the PDBQT file, through the construction of the Vinardo view of the molecules, to atom-pair preprocessing and the scoring stage. During development, smina was used as the reference implementation to compare atom typing, pair preprocessing, and scoring values. The final integration of the components described in this section is collected in PR #105.
PDBQT data and topological representation of the ligand
The first required modification concerned the management of the topological information in the PDBQT file. The standard molecule representation contains atoms, coordinates, atom types, and bonds, but reproducing the behaviour of smina also requires information related to the ligand torsion tree.
In particular, two pieces of data are needed:
- the mobility matrix, used to determine which pairs of ligand atoms can move relative to each other;
- the list of rotors, used to calculate the number of rotatable torsions .
These data are derived from the ROOT/BRANCH/ENDBRANCH structure of the PDBQT file. When a ligand in PDBQT format is parsed, an intermediate representation of the torsion tree is constructed. The ROOT node contains the atoms of the main rigid block, while each BRANCH represents a rotatable bond between a from atom and a to atom, together with the subtree of atoms that depends on that bond.
This representation is used during parsing to construct the mobility matrix with semantics consistent with smina and to obtain the list of ligand rotors. Only the data required by subsequent phases are kept in static_molecule, through the pdbqt_ligand_data field.
This choice is forced by the existing pipeline, where ligands processed in batches are represented as static_molecule, while the protein is loaded separately as dynamic_molecule, stable data for the run. A possible refactor of the type used by the ligand queue remains open. If the pipeline supported a more specific type, the PDBQT data could be moved out of static_molecule and collected in a layer dedicated to Vinardo.
This pragmatic solution avoids reparsing the PDBQT file inside the scoring stage. The stage can instead directly use the mobility matrix and rotor list during batch preparation.
Vinardo layer and atom typing
Once the molecule representation is available, a view of the atoms consistent with the Vinardo scoring model must be constructed. The parsed molecule contains atom types following the AutoDock scheme, while Vinardo uses a set of atom types and properties derived from the model used by Vina.
For this reason, a vinardo_layer was introduced on top of the existing molecule, following the same approach already used by the other layers in the codebase. The layer adds a Vinardo-specific view, exposing the Vinardo type, atomic radius, and flags required to determine whether each atom can contribute to hydrophobic or hydrogen-bond interactions.
Type conversion takes place in multiple steps. First, the AutoDock type used internally by muDock is normalised into an intermediate domain compatible with the Vinardo conversion table. An initial Vinardo type is assigned from this domain, but it may still be ambiguous. Ambiguous cases are then resolved using the topological context of the molecule.
In particular, the bond graph is used to distinguish atoms that have the same initial type but different properties for scoring purposes, for example based on the presence of heteroatom neighbours or polar hydrogens.
The result of the layer is then used by preprocessing to construct valid atom pairs and precompute the pair properties required by the scoring function.
Atom-pair preprocessing
The scoring function is applied to a set of atom pairs selected in advance. For this reason, a preprocessing phase was introduced that constructs the pairs to evaluate during scoring from the Vinardo layers of the protein and ligand.
Protein-ligand pairs are generated by considering every combination of protein and ligand atoms, excluding pairs that contain hydrogens. The ligand-ligand case requires more work because not every internal pair in the ligand must contribute to the score. First, the ligand bond graph is constructed and a Breadth First Search limited to depth three is performed for each atom. The result is saved in a within_three_bonds matrix, which indicates whether two atoms are reachable within three covalent bonds.
A ligand-ligand pair is therefore retained only when both conditions hold: the two atoms are not reachable within three covalent bonds and they are marked as relatively mobile in the mobility matrix constructed from the PDBQT torsion tree.
For each selected pair, the indices of the two atoms, the sum of the atomic radii, and two Boolean flags are saved. One flag indicates whether the pair can contribute to the hydrophobic term and the other whether it can contribute to the hydrogen-bond term. These data are independent from the specific pose and can therefore be calculated once before kernel execution.
In the current implementation, these data are kept in the private buffers of the stage for the current batch. One possible future optimisation would expose them in the pipeline scratchpad if they needed to be reused by multiple stages or preserved beyond a single stage preparation. During genetic exploration, the generated poses change, but ligand topology, atom types, and the set of valid pairs do not. The kernel therefore only needs to update the transformed coordinates and geometric distances, reusing the already computed pair indices, radius sums, and interaction flags.
In general, this preprocessing phase reduces the work performed during scoring. The kernel does not need to query the chemical structure of the molecule and can instead iterate over prepared arrays, calculating only the geometric distances and the energy terms that depend on them for each pose.
Vinardo scoring stage
The central point of the integration is the scoring stage, where the chemical representation built in the previous phases is transformed into the data actually used to calculate the score.
Like other existing stages, the vinardo_score stage implements the three-phase contract. In prepare(), it reads the data available in the scratchpads and constructs the buffers required by the kernel. In operator(), the computation is executed and the SCORES buffer is populated. In teardown(), the result is retrieved and written into the ligand properties.
The stage contains different categories of data with different roles and lifetimes in the computation. They are organised into three levels.
The first level is the device scratchpad, maintained in the stage code as device_scratch and used for data that remains stable across batches. For Vinardo, it contains the protein coordinates in the PROT_X, PROT_Y, and PROT_Z buffers because the protein is shared by every ligand processed during the run.
The second level is the pipeline scratchpad, accessible in the stage through the scratch member and shared by stages operating on the same batch. This scratchpad contains flat buffers used as communication points between stages, produced from the C++ objects constructed previously. For example, in the genetic pipeline, the genetic stage produces new poses by modifying the ligand coordinates and writes them into the X_SCRATCH, Y_SCRATCH, and Z_SCRATCH buffers. The scoring stage then reads the already transformed coordinates from those same buffers and writes the results into SCORES.
The third level consists of the private buffers of the vinardo_score stage. These contain data required only by the Vinardo kernel, such as offsets, indices of protein-ligand and ligand-ligand pairs, sums of atomic radii, and interaction flags. These buffers represent a flattened version of the data described through more convenient C++ objects in the previous phases. The stage converts them into contiguous arrays that are more suitable for sequential kernel access.
The prepare() phase is where this data organisation is concretely constructed for the current batch. It receives a batch of static_molecule objects and first loads data shared with the other stages into scratch, such as atom counts and the working coordinate buffers of the ligands.
Next, a vinardo_ligand is constructed for every ligand in the batch. This object uses the vinardo_layer and the PDBQT data already associated with the molecule to calculate ligand-ligand pairs and the number of rotatable torsions . Protein-ligand pairs are instead generated inside the stage.
The prepare() phase ends by moving the private stage buffers to the device and constructing the vinardo_score_kernel, which receives pointers to those buffers. From this point onward, the computation uses only the linearised data prepared for the current batch.
The operator() phase simply invokes the kernel and delegates the actual score computation to it.
During teardown(), the stage copies the SCORES buffer from the device to the host and assigns the score to the SCORE property of the ligand. In this way, the result reenters the normal output mechanism of the pipeline.
Vinardo scoring kernel
The kernel is the component that consumes the buffers produced by prepare() and calculates the score of the poses in the batch when the stage invokes operator(). The general interface is defined by vinardo_score_kernel<queue_type>. The object maintains pointers to ligand coordinates, protein coordinates, pair arrays, interaction flags, the number of rotatable torsions, and the SCORES buffer.
In this project, the kernel was implemented only for the CPU backend through queue_cpp. In this version, operator() calls calc_vinardo_energy, passing the pointers to the buffers prepared by the stage.
The calc_vinardo_energy() function iterates over the ligands in the batch and the poses associated with each ligand. For each pose, it separately calculates the protein-ligand contribution and the ligand-ligand contribution. For each pair, it reads the atom indices, retrieves the coordinates, and calculates the Euclidean distance between the two atoms. If the distance is greater than or equal to 8 Angstrom, the pair is discarded. Otherwise, the surface distance is calculated by subtracting the precomputed sum of atomic radii from the Euclidean distance. The surface distance and the precomputed hydrophobic and hydrogen-bond interaction flags are then passed to compute_vinardo_pair_energy().
The function that calculates the energy contribution of a single pair was kept separate from the kernel. This choice made it possible to validate the Vinardo formula independently of the execution infrastructure. The separation also makes it possible to expose debugging APIs, useful for observing the individual energy terms and verifying them against the reference.
The kernel keeps the intermolecular and intramolecular contributions separate. This is required to calculate affinity correctly when multiple poses are present for the same ligand. First, the best pose is identified according to the raw score. Each pose is then corrected using the intramolecular contribution of the reference pose and normalised through . The final value is written into the SCORES buffer.
Scoring and genetic pipelines
The pipeline integration is completed in pipeline.hpp, where the vinardo_score_pipeline and genetic_vinardo_pipeline aliases were added. The first instantiates the vinardo_score stage in the scoring-only pipeline, while the second uses it as the scoring stage inside the genetic pipeline. In this way, Vinardo follows the same pattern already used by AutoDock without requiring a separate flow.
The search algorithm and scoring function can be selected at runtime through the CLI options --search, with the values none and genetic, and --score, with the values adt and vinardo. This makes it possible to freely combine the four available configurations.
Validation and Results
This section describes the process used to validate the Vinardo implementation and the results obtained from comparison with the reference.
Validation strategy
Validation was organised incrementally, using smina as the reference implementation for the expected Vinardo behaviour. The final affinity value was therefore not the only value validated. The work was instead divided into the main components of the pipeline: atom typing, pair preprocessing, reconstruction of ligand topology information, and affinity calculation.
To avoid introducing debugging code into the final PR, part of the work was carried out on parallel validation branches. These branches were derived from the corresponding development branches and preserved the same implementation logic, but added dump and instrumentation tools to export data that could be compared with smina. This made it possible to associate atoms, atom pairs, and energy contributions through consistent indices across the two codebases, identifying bugs and divergences at the level of individual components. Development therefore moved to the next component only after the newly introduced one had been completely aligned with the reference.
Validation dataset
During development, a set of 15 complexes from PDBbind v2024 was used to provide broader validation. The considered complexes were identified by the following IDs: 1BNM, 1FKF, 1NLO, 1NLP, 1ABT, 1B2I, 1AO8, 1CWB, 1CWC, 4SGA, 1STD, 220L, 1ILQ, 1B56, and 1EVH.
Incremental validation of the components
The following table summarises the main validation points used during development. For each component, the output produced by muDock was compared with the output obtained from the reference.
| Component | Comparison | Result | Reference |
|---|---|---|---|
| Atom typing | Vinardo types assigned to protein and ligand atoms | Aligned with smina after correction | PR #5 / PR #8 |
| Protein-ligand preprocessing | Protein-ligand atom pairs selected for scoring | Aligned after excluding hydrogens | PR #6 / PR #7 |
| Ligand-ligand preprocessing | Internal ligand pairs selected through topological constraints and relative mobility | Aligned using the PDBQT torsion tree | PR #6 / PR #10 |
| Number of rotatable torsions | used to normalise affinity | Reconstructed as in the reference | PR #10 |
| Scoring | Final affinity value on benchmark cases | Close to the reference, with an explainable residual delta | PR #12 / PR #13 |
Divergences found during validation
The incremental comparison showed that alignment did not depend only on the formula in the paper, but also on several implementation choices. The most relevant cases are reported below because they led to corrections or implementation decisions in the final code.
Atom typing
The per-atom comparison with smina highlighted errors in the table of data associated with the Vinardo types, in particular in the flags and values later used by preprocessing and the scoring function. The table was corrected and the comparison repeated until the same typing as the reference was obtained.
Hydrogen management
In the first preprocessing comparison, muDock generated more atom pairs than smina. The difference came from the reference excluding pairs that contain at least one hydrogen atom, while the initial implementation retained them. Preprocessing was therefore modified to apply the same filter to both protein-ligand and ligand-ligand pairs.
Ligand-ligand pairs
After excluding hydrogens, a divergence remained in the internal ligand pairs. The problem was not related to the topological distance between atoms, but to the definition of relative mobility.
As explained in this comment, the initial implementation used a component already present in muDock: fragments. For each rotatable bond, this component temporarily removes the corresponding connection from the graph, assuming that the rotatable bond represents a valid cut of the graph. A BFS is then performed over the components reachable from the two endpoints of the bond, identifying the atoms on both sides of the cut and constructing the mobile-fragment mask.
This concept was not semantically identical to the one used by the reference, which derives relative mobility from the PDBQT torsion tree. The concrete divergence mainly concerned pairs involving the atoms at the endpoints of the rotor. In the semantics reconstructed from the reference, the from/to pair and the pairs between each of these two atoms and the atoms in the sub-branch are marked as not relatively mobile. The fragment-based logic instead included these pairs among the mobile ones. As a result, preprocessing selected more ligand-ligand pairs than the reference.
To align the behaviour, a mobility matrix with semantics consistent with smina was introduced.
Number of rotatable torsions
Another divergence concerned the value of used to normalise affinity. Here too, directly using either the number of rotors already available in the molecule or the value present in the PDBQT file was not sufficient because the reference applies its own semantics to the rotors that are actually counted.
To align the behaviour, the torsion-tree representation introduced for the mobility matrix was reused. Starting from the rotors described in the PDBQT file, those where at least one endpoint is a hydrogen or does not have at least two bonds with non-hydrogen atoms are excluded. The resulting value is then used in the affinity calculation, making the normalisation consistent with smina.
Curl of positive energies
After aligning the previous components, a residual delta from smina remained. This delta was traced back to curl, a transformation applied by the reference to positive energy contributions before the final normalisation of affinity.
However, curl is applied to intermediate computation results at a level that is not present in the implementation developed in this project. It is also an implementation choice of the reference rather than a term described in the theoretical formulation of Vinardo. For this reason, it was not included in the final implementation.
A test was still performed on an instrumented branch, restructuring the code to expose the same intermediate results. When curl was also applied, the final affinity value aligned with the reference up to machine precision.
Final validation
Final validation on the dataset compared the affinity calculated by muDock using Vinardo with the affinity produced by smina on the same inputs. The results are shown below.
| PDB ID | muDock affinity | smina affinity | Absolute delta |
|---|---|---|---|
| 1BNM | -6.669172 | -6.669182 | |
| 1FKF | -11.700834 | -11.700848 | |
| 1NLO | 1.007401 | 0.992802 | |
| 1NLP | -3.374581 | -3.378935 | |
| 1ABT | -8.456276 | -8.456291 | |
| 1B2I | -2.721796 | -2.723378 | |
| 1AO8 | -6.664783 | -6.664826 | |
| 1CWB | -7.065887 | -7.065935 | |
| 1CWC | -7.466530 | -7.466600 | |
| 4SGA | -8.104091 | -8.104555 | |
| 1STD | -9.236320 | -9.236327 | |
| 220L | -4.433152 | -4.433151 | |
| 1ILQ | -5.019547 | -5.020099 | |
| 1B56 | -5.262350 | -5.262351 | |
| 1EVH | -7.126778 | -7.126916 |
Across the 15 cases, the mean absolute delta was , with a maximum delta of . These results show that the implementation is aligned with the reference, with residual differences explained by the absence of curl in this implementation.
CASF forward-screening benchmark
In addition to numerical validation against smina, a functional forward-screening experiment was performed on the CASF-2016 dataset. The Vinardo test was conducted on the 57 targets of the CASF-2016 benchmark, using the scores produced by the CPU scoring-only pipeline and evaluating them with the official forward-screening script provided with CASF.
In the forward-screening test, 285 ligands and 100 poses per ligand are considered for each target. Each ligand is assigned the best score among its poses and the ligands are ranked according to that score. The top , , and are then evaluated. With 285 ligands and CASF rounding, these correspond to 3, 14, and 29 ligands respectively.
Let be the set of targets, the ligands selected in the top for target , the set of known binders for that target, and the binder with the best experimental affinity. Two metrics were reported:
Success Rate measures the percentage of targets for which the best experimental binder appears in the top . Enrichment Factor measures how much the ranking enriches true binders compared with random selection:
An value close to 1 indicates approximately random behaviour. The denominator represents the expected number of known binders obtained through random selection.
| Method | Ranking | Top 1% (3) | Top 5% (14) | Top 10% (29) |
|---|---|---|---|---|
| Vinardo | lower is better | 33.3% / 9.34 | 49.1% / 4.18 | 63.2% / 3.19 |
Each cell reports Success Rate / Enrichment Factor.
The results show very good enrichment for Vinardo across the complete benchmark of 57 targets, with the best experimental binder recovered in the top , , and for 19, 28, and 36 targets respectively.
Automated CTests
The extended validation over the 15 PDBbind complexes was used during development, but it was not included directly in the CTests of the final PR. The automatic tests instead use the complexes already present in muDock, avoiding the introduction of new datasets into the codebase.
For each complex used in the CTests, a reference affinity value produced by smina was saved. The automatic test runs the muDock Vinardo pipeline on the same PDBQT files and verifies that the calculated affinity remains within an absolute tolerance of from the reference.
In this way, the CTests automatically verify that subsequent changes do not alter the behaviour of the Vinardo pipeline integrated into muDock.
Conclusions
The work introduced the Vinardo scoring function into muDock and integrated it into the existing pipeline. The integration required constructing a Vinardo view of the molecules, converting atom types, preprocessing atom pairs, reconstructing ligand topology information from PDBQT, and implementing the scoring stage with its CPU kernel.
Validation was performed by progressively comparing the components with smina, used as the reference implementation. This made it possible to identify and correct divergences such as hydrogen management, the semantics of relative mobility inside the ligand, and the calculation of the number of rotatable torsions. The final comparison over 15 PDBbind complexes shows good alignment with the reference. The residual differences can be traced back to the curl applied by smina to intermediate contributions, which was not included in the final implementation.
Future developments
Some future developments remain. During PR review, a problem emerged in estimating the memory required by protein-ligand buffers. An initial version restricted Vinardo to a batch size of one because the static estimation API receives only the number of ligand atoms. This constraint was removed by introducing a conservative estimate based on an upper bound for the number of considered protein atoms. The estimate could still be refined, especially for accelerated backends and more precise preallocation.
A second point concerns the auxiliary data extracted from PDBQT. It is currently constructed during static molecule parsing even when it is not required, although it is used only by Vinardo. With runtime selection of the scoring function, this data could be constructed only in the path that uses it.
Another future development is extending the Vinardo kernel to backends other than the CPU version implemented in this project. Finally, parts of preprocessing could be optimised by storing pose-independent data in the scratchpad, such as valid atom pairs and their associated data, avoiding reconstruction when ligand topology does not change.
Conclusion
Overall, the project added a functional Vinardo scoring path to muDock, validated it against an external reference, and integrated it into the stage-based model of the codebase. It provides complete runtime support through the CLI for selecting the search algorithm and scoring function, while leaving a solid foundation for a future port to accelerated backends.
References
- Rodrigo Quiroga and Miguel A. Villarreal, “Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening”, PLOS ONE, 2016.
- Oleg Trott and Arthur J. Olson, “AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading”, Journal of Computational Chemistry, 2010.
- David R. Koes, Matthew P. Baumgartner, and Carlos J. Camacho, “Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise”, Journal of Chemical Information and Modeling, 2013.
- Garrett M. Morris et al., “AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility”, Journal of Computational Chemistry, 2009.
- muDock.
- “Towards High-Performance and Portable Molecular Docking on CPUs through Vectorization”, 2025.